
While the world is facing a new type of biological war after world war-II with corona pandemic caused by coronavirus a piece of positive news struck me two weeks back. New Zealand declares that their country is corona free. With more days in lockdown and staying back at home, communities are fighting on psychological platforms while countries are fighting with their sharp decline in economies. In between all that many countries like South Korea, Taiwan, and New Zealand were able to successfully suppress the first phase of infections and trying to prevent the second phase of COVID-19 pandemic with a very old tool of Biological sciences called “Genomics”.
Scientists around the world are poised to identify the origin of the second wave of infection using Genomics with very advanced tools commonly available in our research institutes. Genome sequencing of first coronavirus strain isolated from Wuhan, China was published on 11th Jan, 20201. Currently, 35000 viral genome sequence of human SARS-CoV2 is available online with a constant increase in their number2. These massive data of viral genome sequencing from different countries:
1) Allows the scientist to understand the origin of the viral outbreak in their own country.
2) Able to identify the genomic variability among different strains may be responsible for a higher or lower rate of pathogenicity.
3) It further helps to contain the spread of the virus by identifying the contact tracing and its geographical origin3.
4) Identifying a specific strain by genomics guide the medical practitioners for precision intervention targeting that particular strain to reduce the spread of infection at a very early stage.
Recently, an analysis published in the journal “Cell” shows a particular mutated variant of the virus is more transmissible compared to other genetic variant4. The genetic variant carrying a mutation in the spike protein region was accounted for just 10% infection in March identified from the sequencing of patients. Dynamic tracking of this coronavirus variant by eminent Joint Consortium of Genomics from Los Alamos National Laboratory the USA and the University of Sheffield, UK identified its global dominance by May accounting for 78% cases around the world. Probably, the mutation in spike protein improves the virus uptake by human cells.
Generally, RNA viruses are prone to mutation and every year we need a new version of the vaccine for flu. Although, the authors in Cell paper did not identify the reason for the high transmissibility of this coronavirus variant among humans the genomic study was successful to identify highly infectious variants. Similarly, identification of mutants which can make virus unrecognizable by our immune system or making it more stable so that it can survive in the air for a longer period will be able to reduce the chances of community spread.
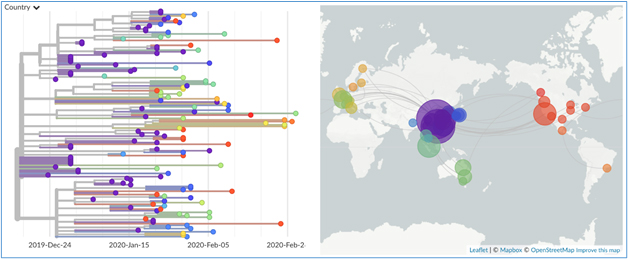
Figure: Nextstrain software platform performs easier and faster representation genomic epidemiology data. The above figure is adopted from Bedford Lab webpage showing the genetic epidemiology of140 novel coronavirus strain (genomic data available at www.gisaid.org) in early March.
During the last decade, genome sequencing techniques have evolved largely due to rapid improvement in technology and operating software. Thanks to Human Genome Project mainly carried out by NIH, the USA in association with other renowned labs around the world completed in 2001-2002 contributed to the rapid development technologies in Genomics. Next-generation sequencing technologies and instruments like Illumina’s Solexa Sequencing or Roche 454 sequencing offered by several companies markedly reduces the time requirements of genome sequencing as well as costs. It is several folds cheaper to do a genome sequencing today in comparison to twenty years back which further opens the gateway for personalized medicine by genome sequencing. Therefore, the requirement for trained peoples in the field of Genomics is rising to operate the instruments, data analysis, and statistical calculations required for a conclusive study.
References:
1.Zhang, Y.-Z. & Holmes, E. C. A Genomic Perspective on the Origin and Emergence of SARS-CoV-2. Cell 2020; 181, 223–227.
3.Stevens, E. et al..The Public Health Impact of a Publically Available, Environmental Database of Microbial Genomes. Front. Microbiol. 2017;8, 808.
4.Korber, B., et al..Tracking changes in SARS-CoV-2 Spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020; https://doi.org/10.1016/j.cell.2020.06.043.
Visited 1892 times, 1 Visit today